Transfusion et maladies du globule rouge
Contacts
Tél.: +33-(0)1 56 72 76 00
cmYuZXRuYXMuc2ZlQGVubmVyaXAuZWNuYXJm
Our team, created in January 2015, is composed of 3 former entities: Etablissement Français du Sang (EFS), Inserm and UPEC. Our 3 entities are involved in red blood cell (RBC) disease pathophysiology and treatment, with a specific focus on sickle cell disease (SCD) and thalassemia. All 3 entities already worked with the referral center of genetic disease of RBC in the H. Mondor Hospital
The former entities are:
– The EFS team directed by Pr F. Pirenne (team leader of the Eq2), which has developed, since 2008 in the H.Mondor Hospital, research projects on transfusion risks in SCD patients and antibody mediated hemolysis in autoimmune haemolytic anemia.
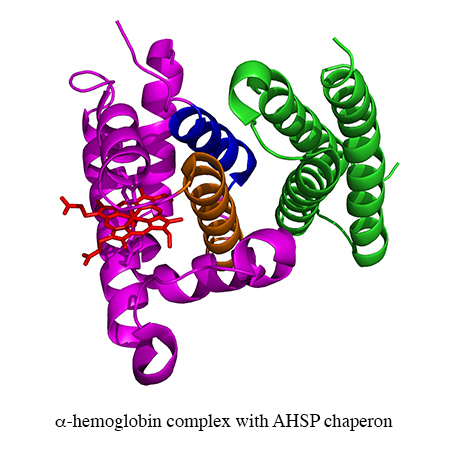
– The Inserm team (ex Inserm U779) specialized in protein polymerisation, blood substitutes and rare RBC diseases. The projects of this team have historically been centred on the RBC, with much of the experimental work at the protein level. The themes include hemoglobin (Hb)-based blood substitutes, the Alpha-Hemoglobin Stabilizing Protein (AHSP), chaperone of alpha chains, and neuroglobin (Ngb).
– The UPEC team specialized in RBC genetic and physiopathology SCD.
The new projects of our group combine the expertise of all members with a specific focus on :
– mechanism of allo immunization against red blood cells
– pathophysiology of post-transfusion hemolysis in sickle cell disease
– role of dense red blood cells in the pathophysiology of sickle cell disease
– study of the free pool alpha in thalassemia and other red blood cell disease
Epidemiological, transversal and fundamental studies are developed on thesis themes.
Our team is a member of the Laboratoire d’Excellence du Globule Rouge, GR-Ex (http://www.labex-grex.com)

Publications récentes
Pirenne F and Yazdanbakhsh K. How I safely transfuse patients with sickle-cell disease and manage delayed hemolytic transfusion reactions
Blood 2018 131:2773-2781Rakotoson MG, Di Liberto G, Audureau E, Habibi A, Fauroux C, Khorgami S, Hulin A, Loric S, Noizat-Pirenne F, Galacteros F, Bartolucci P. Biological parameters predictive of percent dense red blood cell decrease under hydroxyurea.
Orphanet J Rare Dis. 2015 May 9;10(1):57.Noizat-Pirenne F, Habibi A, Mekontso-Dessap A, Razazi K, Chadebech P, Mahevas M, Vingert B, Bierling P, Galactéros F, Bartolucci P, Michel M. The use of rituximab to prevent severe delayed haemolytic transfusion reaction in immunized patients with sickle cell disease.
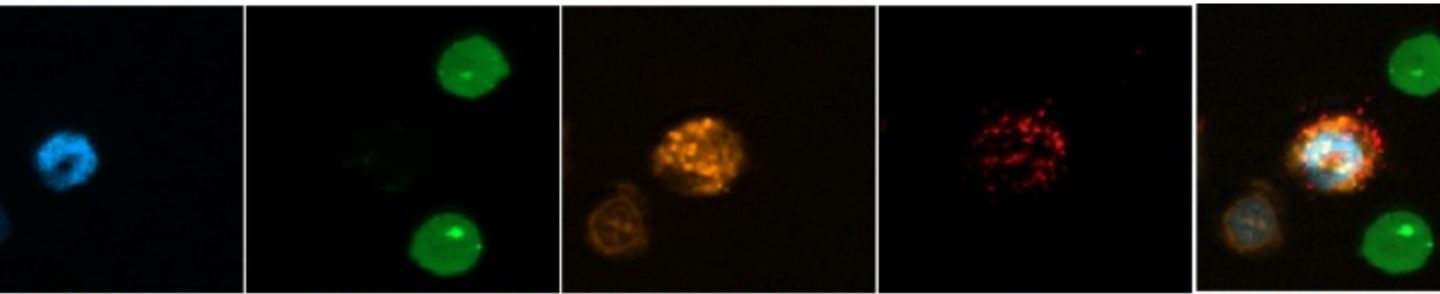
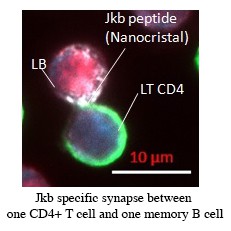
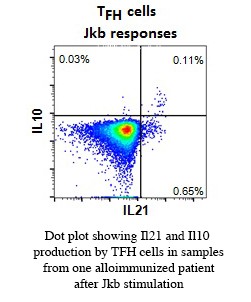
Vox Sang. 2015 Apr;108(3):262-7.Vingert B, Tamagne M, Habibi A, Pakdaman S, Ripa J, Elayeb R, Galacteros F, Bierling P, Ansart-Pirenne H, Bartolucci P, Noizat-Pirenne F. Phenotypic differences of CD4+ T cells in response to red blood cell immunization in transfused sickle cell disease patients.
Eur J Immunol. 2015 Jun;45(6):1868-1879.Burin des Roziers N, Chadebech P, Bodivit G, Guinchard E, Bruneel A, Dupré T, Chevret L, Jugie M, Gallon P, Bierling P, Noizat-Pirenne F. Red blood cell Thomsen-Friedenreich antigen expression and galectin-3 plasma concentrations in Streptococcus pneumoniae-associated hemolytic uremic syndrome and hemolytic anemia.
Transfusion. 2014 Dec 30. doi: 10.1111/trf.12981.Domingues-Hamdi E, Vasseur C, Fournier JB, Marden MC, Wajcman H, Baudin-Creuza V. Role of α-globin H helix in the building of tetrameric human hemoglobin: interaction with α-hemoglobin stabilizing protein (AHSP) and heme molecule.
PLoS One. 2014 Nov 4;9(11):e111395.Arlet JB, Ribeil JA, Guillem F, Negre O, Hazoume A, Marcion G, Beuzard Y, Dussiot M, Moura IC, Demarest S, de Beauchêne IC, Belaid-Choucair Z, Sevin M, Maciel TT, Auclair C, Leboulch P, Chretien S, Tchertanov L, Baudin-Creuza V, Seigneuric R, Fontenay M, Garrido C, Hermine O, Courtois G. HSP70 sequestration by free α-globin promotes ineffective erythropoiesis in β-thalassaemia.
Nature. 2014 Oct 9;514(7521):242-6.Silvy M, Tournamille C, Babinet J, Pakdaman S, Cohen S, Chiaroni J, Galactéros F, Bierling P, Bailly P, Noizat-Pirenne F. Red blood cell immunization in sickle cell disease: evidence of a large responder group and a low rate of anti-Rh linked to partial Rh phenotype.
Haematologica. 2014 Jul;99(7):e115-7.Mellerio C, Farhat WH, Calvet D, Oppenheim C, Lefaucheur JP, Bartolucci P. Cerebral reorganization of language and motor control secondary to chronic hemispheric vasculopathy in a patient with homozygous sickle-cell disease.
Am J Hematol. 2014 Jun;89(6):662-3.Vingert B, Tamagne M, Desmarets M, Pakdaman S, Elayeb R, Habibi A, Bernaudin F, Galacteros F, Bierling P, Noizat-Pirenne F, Cohen JL. Partial dysfunction of Treg activation in sickle cell disease.
Am J Hematol. 2014 Mar;89(3):261-6.Documentation
L'équipe
 COMPOSITION
COMPOSITION
- Responsable de l'équipe :France Pirenne
- Chercheur :Véronique Baudin-Creuza, Pascal Chappert, Nicolas Hebert, Kim-Anh Nguyen-Peyre, Christophe Tournamille, Benoit Vingert
- Enseignant-Chercheur :Pablo Bartolucci, Aline Floc'h, Frédéric Galacteros, Anoosha Habibi, Mehdi Khellaf, Matthieu Mahevas, France Pirenne, Serge Pissard, Mathieu Uzzan
- Praticien hospitalier :Abdourahim Chamouine, Etienne Crickx, Stéphane Moutereau
- Post doctorant :Sevde-Nur Karatas, Deborah Neyrinck-Leglantier
- Doctorant :Saskia Eckert, Christian Kassasseya
- Ingénieur :Emmanuel Adu, Imane Azzaoui, Gwellaouen Bodivit, Elisa Domingues-Hamdi, Sadaf Pakdaman, Marion Pinheiro, Marie Tamagne, Rémy Tristant, Alexis Vandenberghe, Corinne Vasseur
- Technicien :Aurélie Barrault, Nyx Helfman, Alicia Jouard
- Stagiaire :Marion Deret, Sofiane Driouche, Marina Farag, Anaelle Vigne-Haddad
Adresse
IMRB – Inserm U955
Transfusion et maladies du globule rouge (Equipe 2)
Unité d’Ingénierie et de Thérapie Cellulaire Ile-de-France
Etablissement Français du Sang
5, rue Gustave Eiffel
94017 Créteil Cédex
Contact
Tél. : 01 49 81 37 71
Plan d'accès